
FOP
A Fibrodisplasia Ossificante Progressiva (FOP) é uma displasia óssea ultra rara e debilitante em que tecido muscular e tecido conjuntivo (como tendões e ligamentos) são gradualmente substituídos por osso extra-esquelético. Julga-se que a FOP ocorre em cerca de 1 em cada 1,5 milhões de pessoas em todo o mundo1.
O que é a FOP1?
A FOP, também chamada doença de Münchmeyer, é uma doença genética extremamente rara em que o tecido conjuntivo fibroso, como o músculo, tendões e ligamentos, se transforma em tecido ósseo. Sendo a única condição médica conhecida em que um sistema orgânico se transforma noutro. É uma doença grave, incapacitante, sem cura ou tratamento actual.
Esta nova formação óssea (conhecida como ossificação heterotópica) acaba por formar um "esqueleto secundário" que aprisiona as pessoas e lhes restringe progressivamente a capacidade de movimento. O osso formado como resultado deste processo é idêntico ao osso "normal", simplesmente surge em locais impróprios.
Trata-se de uma condição autossómica dominante. Ou seja, uma criança tem 50% de possibiliade de apresentar a doença se um dos seus progenitores for portador. No entanto, na maioria dos casos dois progenitores sem historial da doença podem ter um filho ou filha com FOP, como resultado de uma mutação espontânea (de novo).
O que causa a FOP1,2?
A FOP é causada por mutações no gene ACVR1. Este gene fornece instruções para produzir uma proteína (BMP) que ajuda a controlar o crescimento e desenvolvimento de ossos e músculos, nomeadamente a substituição gradual da cartilagem por osso (ossificação) que ocorre na maturação esquelética normal, desde o nascimento até à idade adulta jovem.
As mutações neste gene vão afetar os mecanismos que controlam o crescimento ósseo e, como resultado, provocam uma ossificação excessiva, característica distintiva FOP.
Características Clinicas1,3
O processo de ossificação heterotópica começa no útero e torna-se geralmente perceptível na primeira infância, começando pelo pescoço e ombros e prosseguindo para os membros. Conheça as características clínicas:
- Metatarsus adductus: metatarsos desviados, ou seja, os ossos na parte dianteira do pé curvam-se ou voltam-se para dentro
- Dificuldade em respirar como resultado da formação óssea à volta da caixa torácica que restringe a expansão dos pulmões
- Tendinites
- Quedas frequentes
- Subutrição devido à incapacidade de abrir completamente a boca que pode causar dificuldade em comer
- Miosite (inflamação muscular) causada por traumas nos músculos. Quedas ou procedimentos médicos invasivos podem provocar edemas e inflamação muscular, seguidos de uma ossificação mais rápida na área lesada
- Ossificação heterotópica
- Polegar hipoplásico, ou seja, redução congénita do tamanho do polegar
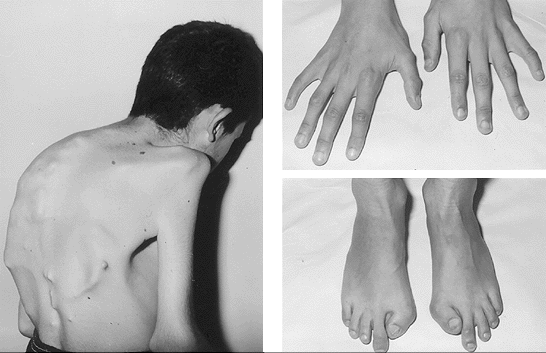
Fig.1. Vista dorsal de rapaz de 13 anos com (esquerda); Clinodactilia dos dedos da mão e hálux (polegar) valgus bilateral curto nos pés.
Fonte: Scielo
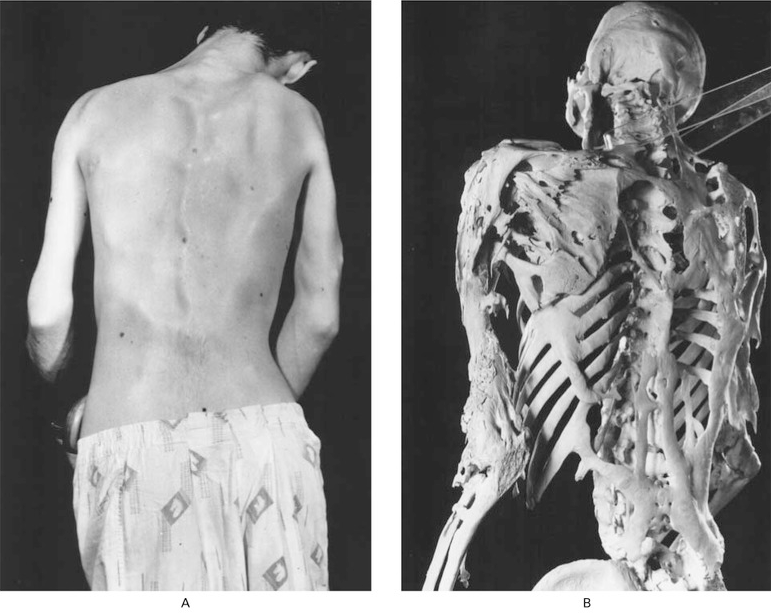
Fig.2. Postura rígida de homem de 25 anos com FOP. Placas de osso ectópico contornam a pele (A) e podem ser vistas directamente no esqueleto (B).
Fonte: Museu Mütter.
Diagnóstico4?
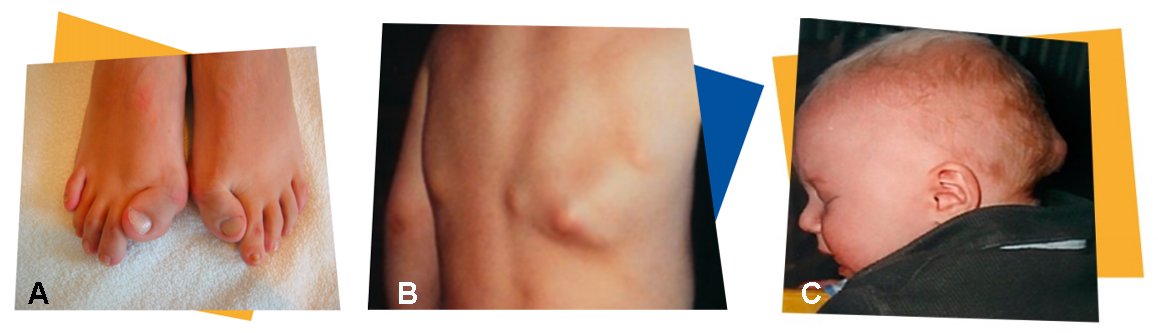
Fig.3. Características observadas na FOP.
Fonte: IPSEN
O desenvolvimento excessivo do osso começa geralmente durante a primeira infância, mas em alguns casos pode apenas dar-se no final da adolescência ou início da idade adulta. Por este motivo, o diagnóstico incorrecto da FOP é comum, mas pode ser evitado examinando os dedos dos pés do bebé em função da característica típica da FOP: hálux (dedo grande) curto e virado para dentro (Fig.3 A) e da identificação da formação óssea extra em locais específicos do corpo (B e C).
Depois de uma avaliação clínica minuciosa, o diagnóstico pode ser confirmado pela sequenciação do gene ACVR1. É de enorme importância referir que os danos latrogénicos (causados por procedimentos médicos), resultantes de falhas de diagnóstico para esta doença rara, são frequentes.
Tratamento5,6
Não existe cura ou tratamento aprovado para a FOP.
Tentativas de remoção cirúrgica de osso, realização de biópsias, injeções intramusculares e de anestésicos locais (terapia dentária) numa pessoa com FOP podem resultar num crescimento exponencial de novo osso nesse local e devem ser evitadas. Enquanto submetidas a anestesia, pessoas com FOP podem encontrar dificuldades na intubação e ter complicações ligadas a doenças pulmonares restritivas e alterações no sistema de condução eléctrica do coração. Atividades que aumentem o risco de queda ou lesão de tecidos moles devem ser evitadas, uma vez que mesmo traumas menores podem provocar a formação de osso heterotópico.
Doenças virais, incluindo a gripe e doenças semelhantes, podem provocar episódios da doença. Nas pessoas com maior susceptibilidade a infeções respiratórias podem ser tomadas medidas preventivas, tais como a terapia antibiótica preventiva (profiláctica).
Sapatos especiais, aparelhos e outros dispositivos que ajudam a caminhar e a suportar o peso têm sido utilizados para ajudar as pessoas com FOP. Deve-se falar com os médicos no sentido de obter este tipo de apoios e dispositivos ou ferramentas especiais para ajudar nas atividades diárias. A terapia ocupacional pode ser benéfica
O aconselhamento genético pode ser benéfico para as famílias com historial de FOP.
Tratamentos em Investigação:
Alguns tipos de medicamentos têm sido utilizados para aliviar a dor e o inchaço associados à FOP durante os surtos agudos (sobretudo corticosteróides) e medicamentos anti-inflamatórios não esteróides entre os surtos. Contudo, estudos mostraram que, apesar de diminuir a inflamação, este tipo de abordagem não diminui a formação de osso.
O Palovarotene é um medicamento desenvolvido como tratamento para doenças ósseas debilitantes ultra-raras e raras. Encontra-se a decorrer um ensaio clínico de Fase 3 de avaliação da eficácia deste medicamento no tratamento da FOP. Consulte a página dedicada aos Medicamentos Orfãos para a FOP, aqui.
O Garetosmab (REGN2477) é outro medicamento que se encontra em estudo para a FOP:
- Um ensaio clínico de Fase 3 com 6 adultos, no Japão, iniciou em outubro de 2021 e analisará a eficácia e segurança deste medicamento. Saiba mais aqui.
- Um ensaio clínico de Fase 2 com 44 adultos (Europa e EUA) para examinar a segurança, tolerabilidade e efeitos do REGN2477 na formação óssea em pessoas com FOP. Saiba mais aqui.
Saiba Mais
- Sobre o medicamento orfão palovarotene para a FOP
- Sobre o processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
- Sobre outros tratamentos para displasias ósseas que se encontram atualmente em estudo
- Sobre doenças raras, displasias ósseas e FOP através dos nossos vídeos informativos
Bibliografia
- 1. Smith, R., Athanasou, N. A., Vipond, S. E. Fibrodysplasia (myositis) ossificans progressiva: clinicopathological features and natural history. Quart. J. Med. 89: 445-456, 1996. PubMed: 8758048
- 2. Shore, E.M., Feldman, G.J., Xu, M. et al. The Genetics of Fibrodysplasia Ossificans Progressiva. Clinic Rev Bone Miner Metab 3, 201–204 (2005). https://doi.org/10.1385/BMM:3:3-4:201
- 3. Cohen, R. et al. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva: a study of forty-four patients. J. Bone Joint Surg. 75A: 215-219, 1993. PubMed: 8423182
- 4. Kitterman, J. A., et al. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics 116: e654-e661, 2005. Note: Electronic Article.
- 5. The medical management of fibrodysplasia ossificans progressiva: current treatment considerations.
- 6. Clinicaltrials.gov