
Displasia Campomélica
A displasia campomélica é uma condição muito rara com prevalência no nascimento de, aproximadamente, 1: 300.000. Trata-se de uma displasia óssea que é frequentemente letal no primeiro ano de vida, devido à insuficiência respiratória relacionada com o tamanho do peito muito pequeno e com a hipoplasia traqueobrônquica1.
O que é a Displasia Campomélica1,2?
A displasia campomélica é uma condição genética grave que afeta o desenvolvimento das vias respiratórias, dos pulmões, dos ossos e dos órgãos reprodutores do bebé. É uma displasia óssea que causa baixa estatura envolvendo um crescimento anormal dos ossos e da cartilagem, apresentando curvatura congénita dos ossos tubulares longos, especialmente nas extremidades inferiores. O nome deriva do grego "campo", que significa curvado, e "melia", que significa membro.
Muitos bebés com esta condição morrem devido a problemas respiratórios. As pessoas que sobrevivem podem desenvolver uma curvatura muito acentuada da coluna vertebral (escoliose) e alterações nos ossos do pescoço que comprimem a medula à medida que envelhecem.
O que causa a Displasia Campomélica1?
Está associada a mutações no gene SOX9. As mutações neste gene também estão associadas à inversão sexual 46 XY, com marcada variabilidade no grau de disgénese gonadal entre portadores da mesma mutação2.
É transmitida num padrão autossómico dominante: alguns casos provêm de um progenitor com mutação, enquanto que outros ocorrem como mutação de novo, ou seja, sem historial familiar (ver imagem abaixo).
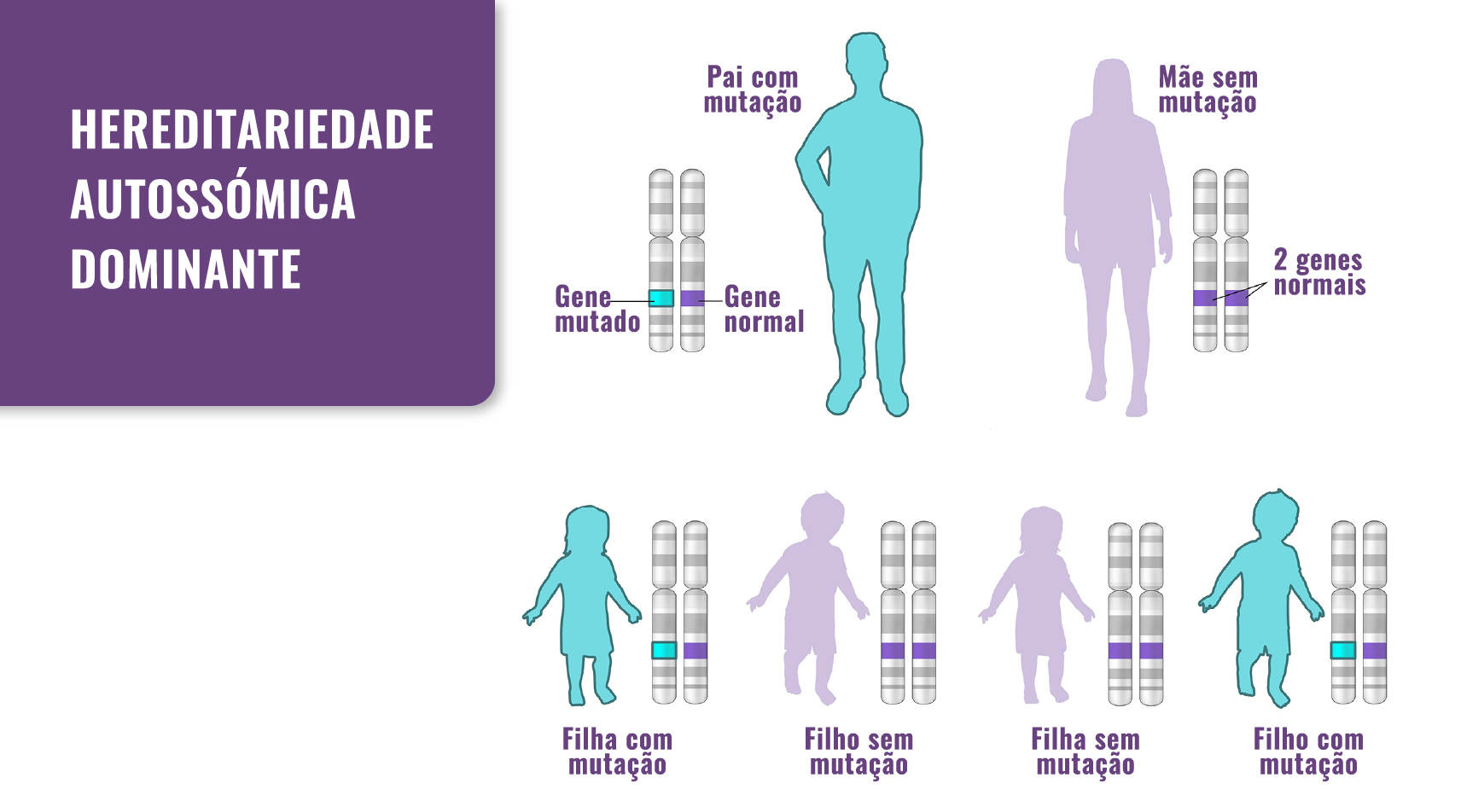
Fig. 1. Hereditariedade: a displasia campomélica passa de pais para filhos. No entanto, também pode ocorrer como resultado de uma mutação de novo.
Características Clínicas e Radiológicas3,4,5

Fig. 2 Características clínicas e radiológicas de bebé aos 7 meses de idade. Fonte: Wiley Online Library
- Dismorfia facial: face com raiz nasal plana, dobras epictáticas, filtrum longo, e micrognatia; macrocefalia e orelhas baixas (A, B)
- Escápulas hipoplásicas, cifose cervical, escoliose torácica com pedículos torácicos não mineralizados, 11 pares de costelas (em vez de 12), isquioses curtas e ramo púbico inferior não fossilizado (C)
- Ossos longos, curvados e frágeis
- Membros inferiores com fémur reto, tíbia e fíbula com ossificação retardada da epífise femoral (D)
- Alargamento da abóbada craniana até ao tamanho dos ossos faciais (E)
- Malformações no peito e pulmões pequenos
- Pé torto
- Metacarpo curto e falanges médias /distais moderadamente curtas (F)
- Ombros mal formados
- Palato fendido com um pequeno maxilar inferior (sequência Pierre-Robin)
- Genitais ambíguos ou genitais femininos normais com um padrão cromossómico masculino típico (46,XY)
- Cartilagem debilitada do trato respiratório superior (laringotraqueomalacia)
- Malformações no cérebro, coração e rins.
Têm sido descritos poucos casos da síndrome variante, referidos como "Displasia campomélica acampomélica". Esta variante pode ser distinguida pela falta de curvatura nos ossos longos, fémures e tíbias3.

Fig. 3. A: Criança de 2 anos com fácies planas típicas, ponte nasal deprimida, queixo pequeno. A laringotraqueomalácia causa deficiência respiratória com traqueostomia; B: Criança de 6 anos com pernas curtas e deformadas. C: Angulação extrema da tíbia com covinha no ápice; D: Pernas curtas e tíbias deformadas em menina de 5 anos: E: Pé torto; metatarso varus. Fonte: Netter.
Diagnóstico2,3
O diagnóstico pré-natal é feito, geralmente, por suspeita durante a ecografia do 2º trimestre, com base na observação de atraso de crescimento ósseo e pseudo-hermafroditismo. O teste genético pré-natal pode ser realizado através da amniocentese ou por biópsia das vilosidades coriónicas.
Após o nascimento, o diagnóstico baseia-se no exame clínico, radiografias das vértebras, ancas, tórax, pernas e pés, ultra-som dos rins e ecocardiograma do coração. A análise do ADN do sangue pode confirmar uma mutação no gene SOX9.
Aconselhamento genético:
O aconselhamento genético é recomendado para ajudar as famílias a compreender a genética e a história natural da displasia campomélica e para fornecer apoio psicossocial.
Efetuar o diagnóstico de uma doença genética ou rara pode constituir muitas vezes um desafio. Os profissionais de saúde normalmente analisam o historial médico e os sintomas, realizam exames físicos e testes laboratoriais, a fim de o alcançar.
Até à data, a maioria das pessoas têm esta condição como resultado de uma mutação de novo. Assim, os pais não têm a condição. Mas o diagnóstico pré-natal de gravidezes de risco acrescido é possível se a variante patogénica na família for conhecida3.
Tratamento3,4,6
Não há tratamento específico para a displasia campomélica. Este destina-se a prevenir ou gerir os sintomas e complicações associados a esta condição. Por exemplo, cuidados ortopédicos e cirurgia podem ser necessários para uma coluna instável, pés tortos, e anomalias da anca. Deverá recorrer-se à cirurgia conforme necessário para minimizar os efeitos da cifose cervicotorácica progressiva na função pulmonar.
A cirurgia também pode ser indicada para o palato fendido, se presente. O tratamento dos problemas respiratórios consiste em assistência respiratória mecânica ou física, tal como pressão expiratória positiva (PEEP).
A Costela Prostética Expansível Vertical em Titânio (VEPTR) foi aprovada pela FDA em 2004 como tratamento para a síndrome da insuficiência torácica em pacientes pediátricos. Trata-se de um dispositivo implantado e expansível que ajuda a endireitar a coluna vertebral e a separar as costelas para que os pulmões possam crescer e encher-se de ar suficiente para respirar.
É essencial a vigilância do crescimento e da curvatura da coluna vertebral e um conjunto variado de especialistas deve estar envolvido nos cuidados e no acompanhamento:
- Médico geneticista
- Ortopedista/cirurgião ortopédico
- Cirurgião craniofacial
- Endocrinologista
- Audiologista
Bibliografia
- 1. Matsushita, M., eta al. A novel SOX9 H169Q mutation in a family with overlapping phenotype of mild campomelic dysplasia and small patella syndrome. Am. J. Med. Genet. 161A: 2528-2534, 2013
- 2. Hsiao HP, et al. Novel SOX9 gene mutation in campomelic dysplasia with autosomal sex reversal. J Formos Med Assoc. 2006 Dec;105(12):1013-6
- 3. Unger S, Scherer G & Superti-Furga A. Campomelic dysplasia. GeneReviews. May 9, 2013
- 4. Jain V, Sen B. Campomelic dysplasia. J Pediatr Orthop B. 2014; 23(5):485-488
- 5. Nelson ME, et al. Campomelic dysplasia: airway management in two patients and an update on clinical-molecular correlations in the head and neck. Ann Otol Rhinol Laryngol. Oct 2011; 120(10):682-5
- 6. Mansour S, et al. The phenotype of survivors of campomelic dysplasia. J Med Genet. 2002; 39:597-602
Saiba mais sobre:
-
O processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
-
A Dor nas displasias ósseas aqui