
XLH
A Hipofosfatémia Ligada ao Cromossoma X — também conhecida por raquitismo hipofosfatémico ou XLH (do inglês X-linked Hipophosphatemia) — ocorre em aproximadamente 3,9-5 bebés por cada 100.000 nascimentos1. É uma displasia óssea rara e genética, caracterizada por baixo nível de fosfato no sangue. Este é expelido em excesso pelos rins, provocando ossos moles e fracos (raquitismo).

O LIVRO DA XLH
Breve viagem sobre uma displasia óssea. Conheça o novo livro da ANDO sobre a XLH
Abrir
SHINE A LIGHT ON XLH
Uma experiência imersiva criada para contar as histórias de pessoas que vivem com XLH
VisitarO que é a XLH1?
É uma condição genética rara com um padrão hereditário dominante ligado ao cromossoma X, embora cerca de 25% dos casos surjam por mutação espontânea. O diagnóstico é feito pelo conjunto do historial familiar, manifestações clínicas e evidências biomédicas.
Os sintomas precoces mais comuns, como as pernas arqueadas ou joelho valgo tornam-se claros quando a criança começa a andar. No entanto, a XLH envolve mais que apenas os ossos e o seu impacto nos indivíduos e famílias pode ser profundo.
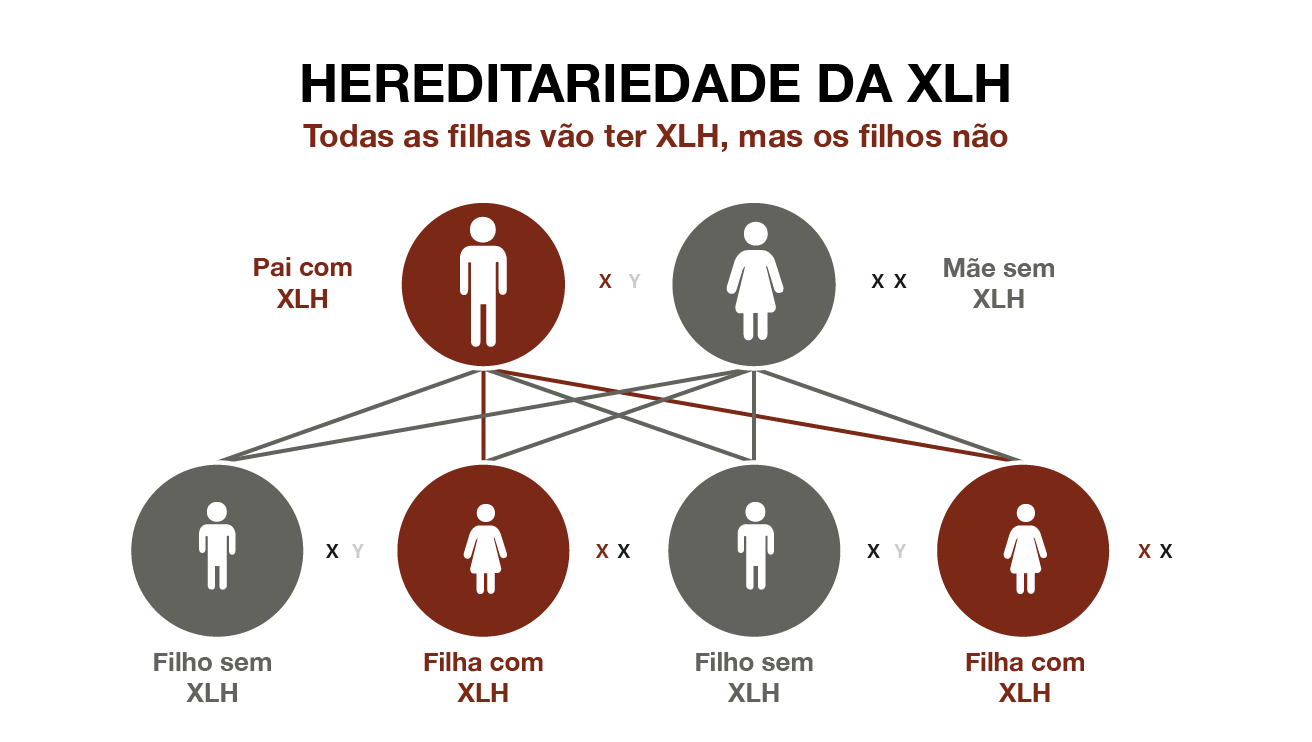
Hereditariedade da XLH
É uma condição genética, passada de pais para filhos, embora cerca de 25% dos casos surjam por mutação espontânea (mutação de novo).
O que causa a XLH1?
A XLH é causada por uma mutação no gene PHEX. Este gene é responsável pela regulação do fosfato no corpo, que leva a que os rins excretem mais fosfato que o normal. Consequentemente, conduz a baixos níveis de fosfato no sangue afetando a mineralização normal dos ossos e dentes2,3. Os desafios começam na infância, durante o crescimento, persistindo e, frequentemente, aumentando durante idade adulta.
Características Clinicas4,5,6
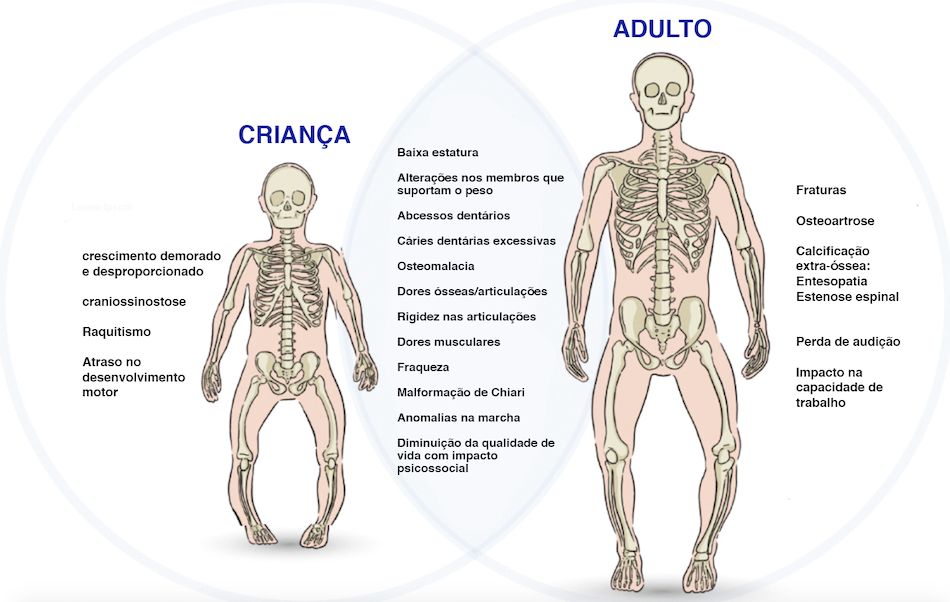
Fig. 1. Sintomatologia e fisiopatologia da XLH. Sinais, sintomas, sequelas e consequências a longo prazo em crianças e adultos.
Fonte: adaptado de Orphanet Journal of Rare Diseases.
Algumas pessoas com XLH não têm sintomas aparentes relacionados com os ossos, apresentando apenas hipofosfatémia, enquanto outras podem ter sintomas graves. Em muitos casos, os sintomas tornam-se aparentes nos primeiros 18 meses de vida, quando a criança começa a ter peso nas pernas:
- Desenvolvimento ósseo anormal (curvatura ou torção da parte inferior das pernas)
- Dor óssea e muscular
- Dor articular causada pelo endurecimento dos tendões e ligamentos
- Fraqueza
- Marcha bamboleante
- Desenvolvimento anormal dos dentes, abcessos dentários e dor
- Raquitismo que não melhora com a terapia tradicional com Vitamina D
- Desenvolvimento anormal do crânio
- Baixa estatura ou taxa de crescimento mais lenta
- Genu varum
Diagnóstico
A XLH é diagnosticada com base num exame físico, imagiologia raio-x, testes sanguíneos e no historial familiar. Os factores específicos considerados para o diagnóstico incluem:
- Uma taxa de crescimento lenta e uma curvatura notável das pernas
- Níveis baixos de fosfato no sangue
- Níveis elevados de FGF23 no sangue
- Falta de resposta do nível de fosfato ao tratamento com vitamina D
- Embora não sejam essenciais para o diagnóstico, os testes genéticos para a XLH podem confirmar a mutação
O Impacto de Viver com XLH

Fig. 2. Outros desafios de viver com XLH.
A diferença física causada pela XLH está associada ao estigma e ao bullying e os desafios psicológicas afetam as crianças e os adultos. Viver com uma condição genética inclui saber que esta pode ser passada para os descendentes, o que causa angústia emocional.
As pessoas com XLH podem precisar de adaptações em casa, no trabalho e na escola e a incapacidade pode tornar-se muito desafiante, levar à depressão e influenciar a habilidade no trabalho, o desempenho na escola e complicar as tarefas do dia-a-dia.
Tratamento
Para determinar a extensão da doença e as necessidades de cada indivíduo são recomendadas as seguintes avaliações:
- Nas crianças:
- Uma radiografia às extremidade inferiores e aos pulsos para avaliar a extensão da doença
- Medição do desenvolvimento ósseo para avaliar o potencial de crescimento
- Exame craniofacial para encntrar sinais de craniossinostose
- Exame dentário
- Avaliação da audição - Nos adultos:
- Radiografias de locais do esqueleto com dor relatada para avaliar entesopatias ou fracturas
- Exame dentário
- Avaliação da audição - Em qualquer idade:
- Avaliação das pessoas com dores de cabeça e vertigens (malformação de Chiari)
- Consulta de aconselhamento genético
São necessários suplementos de fosfato, habitualmente combinados com uma dose elevada de calcitriol, que permite uma melhor absorção do cálcio7. Nas crianças, o tratamento é geralmente iniciado no momento do diagnóstico e continua até que os ossos parem de crescer2. Nos adultos, o principal objectivo do tratamento é atenuar a dor.
Outros tratamentos para XLH, dependendo dos sintomas e da gravidade, podem incluir2,7:
- Hormona de crescimento
- Cirurgia correctiva para fixar pernas arqueadas ou dobradas
- Tratamento para reparar anomalias do crânio, tais como fusão prematura dos ossos do crânio (sinostose)
- Procedimentos dentários para tratar a dor nos dentes e nas gengivas
Tratamento aprovado pela EMA/FDA:
O medicamento Burosumab (comercializado com o nome Crysvita) é utilizado para o tratamento da XLH e pode ser utilizado em crianças com mais de 1 ano de idade, adolescentes com esqueletos em crescimento, caso sejam observados sinais de alteração óssea em radiografias e já se encontra aprovada indicação para adultos também.
A XLH é uma condição rara, por isso o Burosumab recebeu designação de Medicamento Órfão a 15 de Outubro de 2014, pela Agência Europeia do Medicamento (EMA). Uma vez que obteve uma aprovação condicional, a empresa Kyowa Kirin — farmacêutica e biotecnológica japonesa — responsável pela sua comercialização fornecerá resultados atualizados sobre estudos realizados. Consulte informação completa sobre este medicamento aqui 8.
Como melhorar os resultados em saúde na XLH?
-
Profissionais de SaúdeÉ necessário que a comunidade de profissionais de saúde e cuidadores reconheçam que a XLH é incurável, para toda a vida e que está associada a uma série de sintomas debilitantes.
-
DiagnósticoÉ essencial um diagnóstico precoce e preciso de forma a que o tratamento possa ser iniciado prontamente; todos os recém-nascidos com XLH no historial familiar devem ser testados para a presença da mutação.
-
ReferenciaçãoOs médicos de cuidados primários devem ser informados sobre as características desta condição e para a necessidade de referênciação urgente.
-
Acompanhamento multidisciplinarTodos as pessoas com XLH devem ser avaliadas e seguidas por equipas multidisciplinares que incluam serviços médicos e de suporte.
-
Acesso a medicaçãoA disponibilização de medicação deve ser atempada e monetariamente acessível. Esta deve também ser ajustada às necessidades individuais, sem diferenças relativas ao país de residência ou às circunstâncias socioeconómicas.
-
Orientações médicas padronizadasSão necessárias linhas orientadoras internacionais baseadas na investigação e desenvolvidas com participação de pessoas com XLH, de forma a garantir um tratamento consistente a todas as pessoas.
-
Informação adequadaDeve ser fornecida informação confiável e de acesso fácil às pessoas e suas famílias, de forma a evitar que subsistam dúvidas. As pessoas com XLH devem conhecer os seus direitos.
Como posso ajudar?
- Pode apoiar as pessoas com XLH partilhando estas informações do site da ANDO
- Pode contribuir com donativos para as organizações e para a investigação sobre a XLH
- Pode-se voluntariar para apoiar a associação do seu país ou a International XLH Alliance
Saiba Mais
- Sobre o medicamento orfão burosumab para a XLH
- Sobre o processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
- Sobre outros tratamentos para displasias ósseas que se encontram atualmente em estudo
- Sobre doenças raras, displasias ósseas e XLH através dos nossos vídeos informativos

Descarregue este infográfico aqui
Bibliografia
- 1. Davies M, Stanbury S. The rheumatic manifestations of metabolic bone disease. Clin Rheum Dis. 1981;7:595–646
- 2. Ruppe MD. X-Linked Hypophosphatemia. GeneReviews; jan 7, 2021
- 3. Dixon PH, et al. Mutational analysis of PHEX gene in X-linked hypophosphatemia. J Clin Endocrinol Metab. 1998;83:3615–23
- 4. Carpenter TO, et al. A clinician's guide to X-linked hypophosphatemia. J Bone Miner Res. 2011;26:1381–8
- 5. Hardy DC, et al. X-linked hypophosphatemia in adults: prevalence of skeletal radiographic and scintigraphic features. Radiology. 1989;171:403–14
- 6. Pronicka E, et al. Anthropometric characteristics of X-linked hypophosphatemia. Am J Med Genet A. 2004;126A:141–9
- 7. Chan JCM. Hypophosphatemic Rickets. Medscape Reference. jan 7, 2021
- 8. EMA. Crysvita, burosumab. jan 7, 2021. Consultar.
Veja o vídeo "Saiba Mais Sobre XLH"

Conheça a XLH Alliance
A XLH Alliance é uma aliança internacional constituída por mais de 23 organizações de diferentes países, onde se inclui a ANDO. Tem como objetivo amplificar a voz das Pessoas com XLH no mundo e estabelecer um padrão global multidisciplinar de cuidados e investigação.
visitar site
Agradecimentos
À Tenna Toft Silvest — Presidente da Associação Dinamarquesa de XLH e vice-presidente da International XLH Alliance — por ceder a fotografia de topo, tirada a pedido da ANDO e em específico para esta página. A Tenna tem XLH, é uma excelente colega e tem 2 filhas gêmeas de 3 anos, uma também tem XLH.