
Síndrome 3M
A síndrome 3M é uma displasia óssea extremamente rara. Até à data, foram registados cerca de 200 casos desta doença hereditária. No entanto, é provável que o fenótipo esteja sub-reconhecido.
O que é a Síndrome 3M1,2?
A síndrome 3M (OMIM 273750) é uma doença autossómica recessiva que foi descrita pela primeira vez em 1975. Foi designada por 3M mais tarde com base nas iniciais dos 3 primeiros investigadores: Miller, Mckusick e Malvaux. É caracterizada por restrição de crescimento durante e após o período pré-natal, alterações radiológicas, aparência facial distinta, entre outras características como: pescoço curto, macrocefalia relativa, ossos longos finos, corpos vertebrais altos e calcanhares proeminentes (marcador universal). A função endócrina é normal.
O mecanismo molecular na base da síndrome 3M foi descrito em 2005, quando foram identificadas variantes patológicas no gene CUL7 (cullin 7).
O que causa a Síndrome 3M1,2?
A síndrome 3M é herdada como uma caraterística genética autossómica recessiva. As doenças genéticas recessivas ocorrem quando um bebé herda duas cópias de um gene, uma de cada progenitor. Se um bebé receber um gene normal e um gene com mutação, será portador da doença (heterozigoto) e pode apresentar algumas alterações físicas ligeiras (por exemplo, anomalias craniofaciais subtis e/ou ossos invulgarmente delgados).
O risco de dois pais portadores transmitirem o gene mutado e terem um filho afetado é de 25% em cada gravidez. O risco de ter um filho que seja apenas portador é de 50%. A probabilidade de receber genes normais de ambos os pais é de 25%.
Existe heterogeneidade genética na 3M. Atualmente, sabe-se que as mutações num de três genes causam a síndrome 3M: CUL7, OBSL1 e CCDC8. Uma vez que as mutações nos três genes identificados até à data não são responsáveis por 100% dos casos de síndrome 3M, postula-se que podem estar envolvidas mutações de outros genes (potencialmente membros da mesma via).
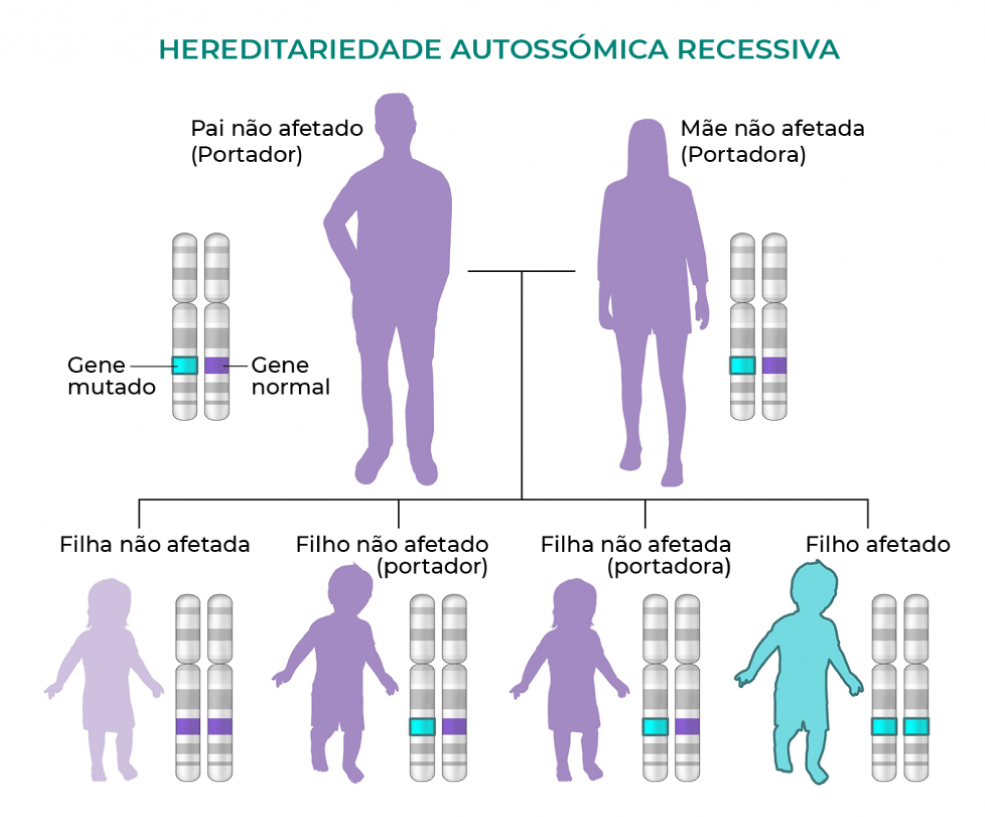
Fig. 1. Hereditariedade autossómica recessiva.
Características Clínicas e Radiológicas2,3,4
Características clínicas
-
Baixa estatura em período pré-natal
-
Dolicocefalia
-
Dismorfismo facial, sendo que a aparência da face varia entre indivíduos afetados
-
Face triangular
-
Macrocefalia relativa
-
Fronte (testa) proeminente
-
Hipoplasia da face média
-
Sobrancelhas grossas e retas
-
Ponta do nariz carnuda
-
Lábios grossos e queixo bicudo
-
Cílios longos
-
Comprimento à nascença entre 40-43cm
-
Anomalias geniturinárias em indivíduos do género masculino como hipogonadismo e hipospadia
Músculo-esqueléticas:
-
Esterno deformado
-
Escápula alada
-
Cifoscoliose torácica
-
Espinha bífida oculta
-
Clinodactilia do 5º dedo
-
Hipermobilidade generalizada ou isolada das articulações
-
Polegares flexíveis
-
Calcanhares proeminentes
-
Pés planos
-
Pescoço largo e curto
-
Músculo trapézio proeminente
Características radiológicas
-
São subtis, por norma aparecem após os dois anos de idade e podem incluir as seguintes:
-
Ossos longos e finos com constrição diafisária e metáfises alargadas.
-
Colos femorais podem ser curtos.
-
Corpos vertebrais são altos, com diâmetro anterior-posterior e transversal reduzido (especialmente na zona lombar)
-
Cunha dos corpos vertebrais torácicos e placas terminais superiores e inferiores irregulares
-
Cifoscoliose torácica
-
Spina bifida oculta
-
Tórax relativamente largo com costelas horizontais delgadas
-
Ossos pélvicos pequenos, especialmente a púbis e o ísquio
-
Asas ilíacas alargadas
-
Foramen Magnun pequeno, embora possa ser posicional.
-
Distância intra-orbital estreita,
-
Displasia do cotovelo
-
Ulna encurtada
-
Pseudoepífises do segundo osso metacarpo
-
Anca deslocada
-
Astrágalo proeminente
-
Sutura coronal achatada.
Heterogeneidade Genética3,4
-
3M-2 (OMIM 612921), causada por mutação no gene OBSL1 no cromossoma 2q35
-
3M-3 (OMIM 614205), causada por mutação no gene CCDC8 no cromossoma 19q13
-
Marcador universal - calcanhar proeminente presente em 3M1, 3M2, 3M3.
Testes genéticos
É recomendado teste genético nos casos em que existem características clínicas e radiológicas específicas da 3M. A identificação de variantes patogénicas bialélicas em CUL7, OBSL1 e CCDC8 podem ajudar a chegar ao diagnóstico, se a avaliação clínica e radiológica for inconclusiva (Fig.1)
 Fig. 1 - Variantes patogénicas bialélicas em CUL7, OBSL1 E CCDC8. Créditos: Florence Barsoum
Fig. 1 - Variantes patogénicas bialélicas em CUL7, OBSL1 E CCDC8. Créditos: Florence Barsoum
É possível serem realizados os seguintes testes genéticos:
-
Sequenciação do exoma
-
Sequenciação do genoma
-
Gene-target (sanger sequencing)
-
Painel de genes para displasias ósseas
Diagnóstico4,5,6,7
O diagnóstico do Síndrome 3M deve ser considerado quando existe um probando, ou seja, a pessoa de uma família na qual é detetada pela primeira vez uma determinada doença ou alteração genética; e/ou quando há combinação de características clínicas e radiológicas referidas acima.
Diagnóstico diferencial
-
Nanismo Mulibrey
-
Syndrome de Silver Russel
-
Syndrome de Dubowitz
-
Syndrome alcoólica fetal
A Síndrome 3M pode ter outras designações tais como:
-
Displasia dolicospondilítica
-
Face tristonha
-
Síndrome de le Merrer
-
Nanismo primordial da síndrome 3M
Tratamento4,5,6,7
Não existe um tratamento específico para a síndrome 3M, contudo, o alongamento ósseo, que consiste numa abordagem cirúrgica para aumento de estatura, pode ser uma opção. O tratamento com hormona de crescimento tem dado resultados distintos entre indivíduos.
Aconselhamento genético familiar
A síndrome 3M é herdada de modo autossómico recessivo. Cada irmão de um probando tem 25% de probabilidade de não ser afetado nem portador, 50% de ser assintomático e portador, e 25% de ser afetado e por isso portador. É aconselhável realizar testes genéticos para famílias em risco ou no período pré-natal é possível para onde se identificou variantes patogénicas num membro afetado. É possível a deteção do abrandamento do crescimentos de todos os ossos por ecografias no pré-natal.
 Fig. 2 - Gráfico estaturo-ponderal de um probando com mutação no gene CUL7
Fig. 2 - Gráfico estaturo-ponderal de um probando com mutação no gene CUL7
Bibliografia
2. Irving M, Holder-Espinasse M. Three M Syndrome. 2002 . In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews
3. Takatani, T., Shiohama, T., Takatani, R. et al. A novel CUL7 mutation in a Japanese patient with 3M syndrome. Hum Genome Var 5, 30 (2018)
4. Hanson D, Murray P, Black G, Clayton P, The Genetics of 3-M Syndrome: Unravelling a Potential New Regulatory Growth Pathway, Horm Res Paediatr 2011;76:369–378
Resumo adaptado de artigo escrito por Florence Barsoum, consultar artigo aqui.
Agradecemos à Florence a partilha da foto do seu filho Guilherme.
Saiba mais sobre:
-
O processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
-
A Dor nas displasias ósseas aqui