
Osteocondromas Múltiplos
Os osteocondromas múltiplos hereditários (OMH) são uma doença genética, rara, gravemente incapacitante, progressiva e crónica que provoca o crescimento de tumores ósseos benignos em crianças e adolescentes. A OMH é tipicamente diagnosticada na primeira infância, quando os tumores se tornam visíveis, normalmente aos quatro anos de idade. A sua prevalência é estimada em 1:50.000 e, aparentemente, é superior nos rapazes (rácio masc-fem: 1,5:1)1.
O que são OMH1,2?
Os osteocondromas múltiplos hereditários (OMH), também conhecidos como ou exostoses múltiplas hereditárias ou Osteocondromatose Múltipla Familiar, são uma doença genética causada por uma mutação num gene. Esta mutação provoca a formação de muitos tumores benignos, conhecidos como osteocondromas (OCs) ou exostoses osteocartilaginosas, em determinados ossos durante a infância e adolescência, até ao fecho das placas de crescimento.
Devido ao seu desenvolvimento em torno das articulações, as crianças desenvolvem deformidade nos membros e apresentam restrição de movimentos à medida que crescem.
O que são osteocondromas?
Os OCs são tumores ósseos benignos que se formam quando as células dos ossos crescem de uma forma atípica. Formam-se na superfície plana dos ossos ou nas placas de crescimento e um pedaço de cartilagem (tecido flexível que faz parte do esqueleto), conhecido como capa de cartilagem, cobre-os.
O número de OCs pode variar significativamente de pessoa para pessoa, sendo o número médio entre 15-18 OCs. A maioria é assintomática e localizada em ossos que se desenvolvem a partir da cartilagem, principalmente nos ossos longos das extremidades, predominantemente ao redor do joelho.
O que causa os OM1?
Os OMH são uma doença genética com padrão hereditário dominante (ver Fig. 1). Em 90% das pessoas com OMH, a doença ocorre quando há uma mutação no gene EXT1 ou no EXT2. Para os outros 10%, a causa genética é ainda desconhecida.
Na maioria das vezes, a doença ocorre quando os pais transmitem estas mutações genéticas aos filhos. No entanto, algumas pessoas com OMH não têm um progenitor com esta mutação genética. Neste caso, sem antecedente familiar, ocorre por mutação de novo.
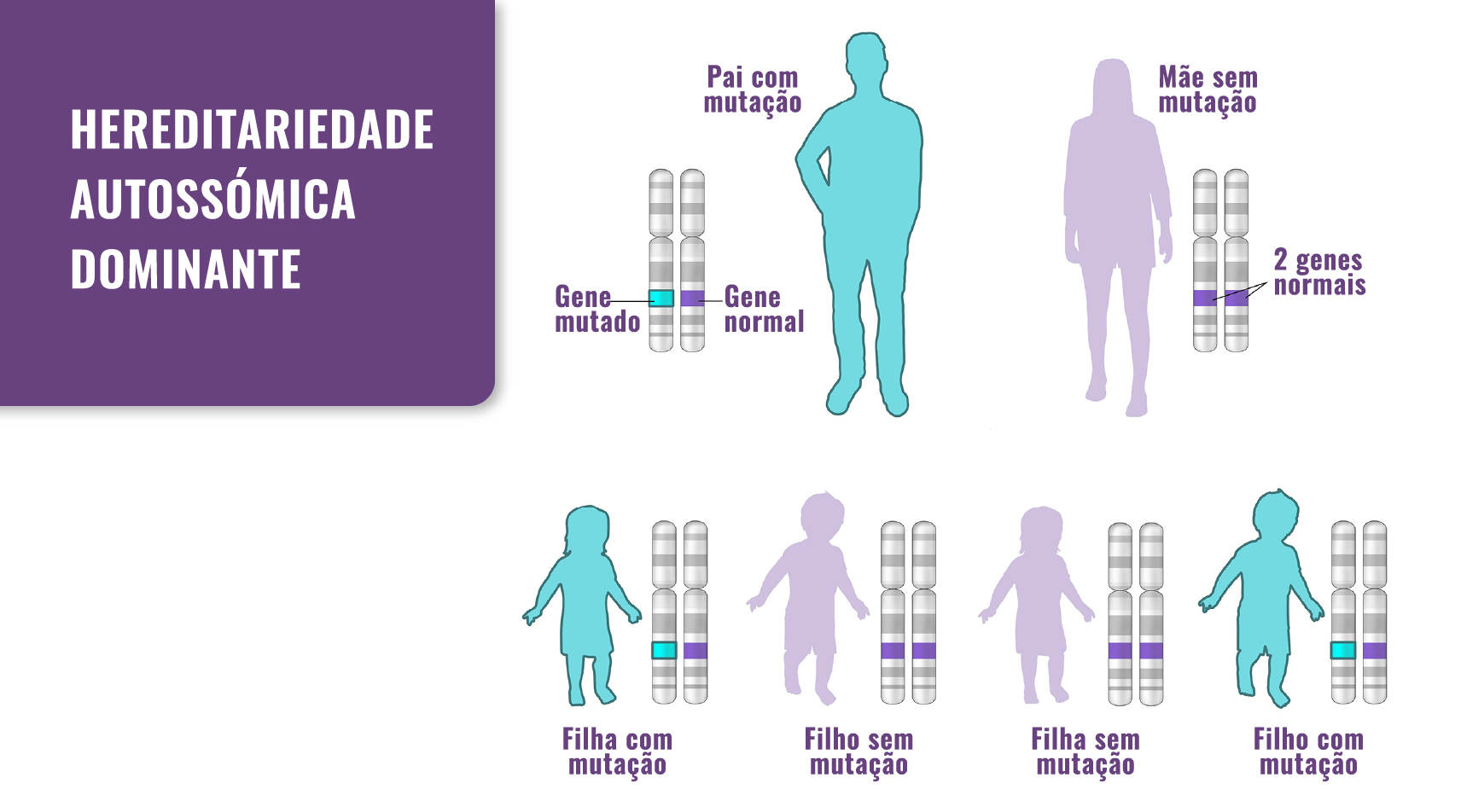
Fig. 1. Hereditariedade: os OMH passam de pais para filhos de forma autossómica dominante.
Características Clínicas e Sintomas3,4,5,6
Os OMH provocam nódulos que podem ser dolorosos num ou mais ossos. Outros sintomas incluem:
-
Crescimento anormal dos ossos, incluindo alterações nos ossos do antebraço
-
Fracturas ósseas e fracturas da placa de crescimento.Discrepância no comprimento dos membros
-
Perda de movimento das articulações
-
Nervos comprimidos
-
Osteoartrite prematura
-
Problemas nos tornozelos ou nos joelhos
-
Dores a curto e a longo prazo
-
Baixa estatura
-
Algumas crianças podem sentir dor se os tumores crescerem perto de vasos sanguíneos ou nervos. Por vezes, os inchaços podem ser indolores
-
Os ossos da face não são afetados
-
Lesões ósseas em ossos chatos, vértebras e costelas são menos comuns e habitualmente não ocorre envolvimento do crânio
-
Os OCs podem causar dor, alterações funcionais e deformidades (especialmente do antebraço), o que pode justificar a sua remoção cirúrgica
-
A complicação mais importante é a transformação maligna do osteocondroma em tumor cartilaginoso atípico periférico secundário ou condrossarcoma, que ocorre em 0,5-5% dos casos.

Figura 2. (A) Película posterior e lateral de um condrossarcoma de grandes dimensões na omoplata direita que foi diagnosticado como um tumor maligno revestido por cartilagem. (B) Imagens clínicas de condrossarcomas no fémur distal bilateral e na tíbia proximal esquerda. (C) Imagens clínicas de condrossarcoma na tíbia distal esquerda.
Wang, W., et al. (2022). Clinical survey of a pedigree with hereditary multiple exostoses and identification of EXT‑2 gene deletion mutation. Molecular Medicine Reports, 25(4).

Figura 3. - (A) Imagens intra-operatórias. Massas da omoplata foram retiradas e biópsia mostrou tumor maligno com capa de cartilagem. (B) Massas da tíbia e do perónio retiradas e biopsia mostrou tecido tumoral lobulado (a), composto por condrócitos (b) e cartilagem (c). (C) No seguimento de dois anos, não houve recorrência de OCs.
Wang, W., et al. (2022). Clinical survey of a pedigree with hereditary multiple exostoses and identification of EXT‑2 gene deletion mutation. Molecular Medicine Reports, 25(4).
Diagnóstico2,3,6
Como é que os OMH são diagnosticados?
O diagnóstico baseia-se na evidência imagiológica e clínica, complementada, se disponível, pela avaliação histológica dos OCs. Os pais ou um profissional de saúde podem notar um nódulo num osso ou perto dele. Os profissionais de saúde podem então recomendar exames imagiológicos como uma radiografia para ver os OCs. Se necessitarem de mais informações, podem recomendar um TAC ou uma ressonância magnética.
Os critérios de diagnóstico (OMS 2020) incluem achados radiológicos de pelo menos dois OCs na região justa-epifisária de ossos longos e história familiar positiva e/ou mutação germinativa comprovada num dos genes EXT.
Se a mutação exata for conhecida, o diagnóstico pré-natal é tecnicamente possível.
Diagnóstico diferencial:
Os OMH devem ser diferenciados da metacondromatose, da displasia epifisária hemimélica, da doença Ollier e de um osteocondroma solitário.
Aconselhamento genético:
O aconselhamento genético é recomendado para ajudar as famílias a compreender a genética e a história natural dos OMH e para fornecer apoio psicossocial.
Os OMH são uma doença autossómica dominante e geneticamente heterogénea. Deverá ser disponibilizado aconselhamento genético a pessoas com a mutação causadora da doença, informando-as de que há 50% de risco de transmissão da mutação aos seus descendentes.
Tratamento1,6
Não existe cura para os OM. Os tratamentos são dirigidos para os sintomas específicos à medida que estes se manifestam e, normalmente, requerem esforços coordenados de uma equipa multidisciplinar.
O seguimento clínico inclui a remoção dos OCs quando sintomáticos. Os OCs removidos devem ser examinados relativamente à transformação maligna em tumor cartilaginoso atípico periférico secundário ou condrossarcoma.
As pessoas devem ser bem instruídas e acompanhadas regularmente para a deteção precoce de malignidade. Para o condrossarcoma periférico secundário, deve ser realizada resseção em bloco da lesão e da sua pseudocápsula com margens livres de tumor, preferencialmente num centro de referência de tumores ósseos.
A gestão das patologias e vários problemas de físicos derivados dos OMH incluem:
Observação do tumor. Controlo da dor. Fisioterapia. Cirurgia para alongar os membros que não cresceram corretamente. Cirurgia para remover o(s) tumor(es).
O acompanhamento psicoemocional relacionado com a dor, deformidades físicas, e baixa estatura é recomendado para para as pessoas com OMH, assim como para as suas famílias.
Tratamentos em Investigação
Um ensaio clínico com o potencial medicamento palovarotene, da farmacêutica IPSEN, para idade inferior a 14 anos foi suspenso em janeiro de 2020, com base nos resultados de uma análise de futilidade.
Bibliografia
- 1. Bovée JVMG, Hogendoorn PCW. Multiple osteochondromas. In: Fletcher CDM, Unni KK, Mertens F, eds. World Health Organization. Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon, France: IARC Press; 2002:360-2.
- 2. Clement ND, Duckworth AD, Baker AD, Porter DE. Skeletal growth patterns in hereditary multiple exostoses: a natural history. J Pediatr Orthop B. 2012;21:150–4.
- 3. Clement ND, Porter DE. Forearm deformity in patients with hereditary multiple exostoses: factors associated with range of motion and radial head dislocation. J Bone Joint Surg Am. 2013;95:1586–92.
- 4. Fei L, Ngoh C, Porter DE. Chondrosarcoma transformation in hereditary multiple exostoses: a systematic review and clinical and cost-effectiveness of a proposed screening model. J Bone Oncol. 2018;13:114–22.
- 5. Hennekam RC. Hereditary multiple exostoses. J Med Genet. 1991;28:262–6.
- 6. Jennes I, Pedrini E, Zuntini M, Mordenti M, Balkassmi S, Asteggiano CG, Casey B, Bakker B, Sangiorgi L, Wuyts W. Multiple osteochondromas: mutation update and description of the multiple osteochondromas mutation database (MOdb). Hum Mutat. 2009;30:1620–7.
Saiba mais sobre:
-
O processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
-
A Dor nas displasias ósseas aqui