
Displasia Diastrófica
A displasia diastrófica é uma displasia óssea rara que afeta a cartilagem e o desenvolvimento ósseo. Embora a prevalência exata desta condição seja desconhecida, estima-se que afeta cerca de 1 em 500.000 recém-nascidos nos Estados Unidos. No entanto, é mais comum na Finlândia, onde afecta cerca de 1 em cada 33.000 bebés2.
O que é a Displasia Diastrófica1,2?

Brody Stanley, um menino de 6 anos com displasia diastrófica, reside no Estado americano de Delaware. Carregue na imagem para conhecer a sua história (em inglês) e siga a aventura do Brody no Instagram aqui. Créditos: Marcella Stanley
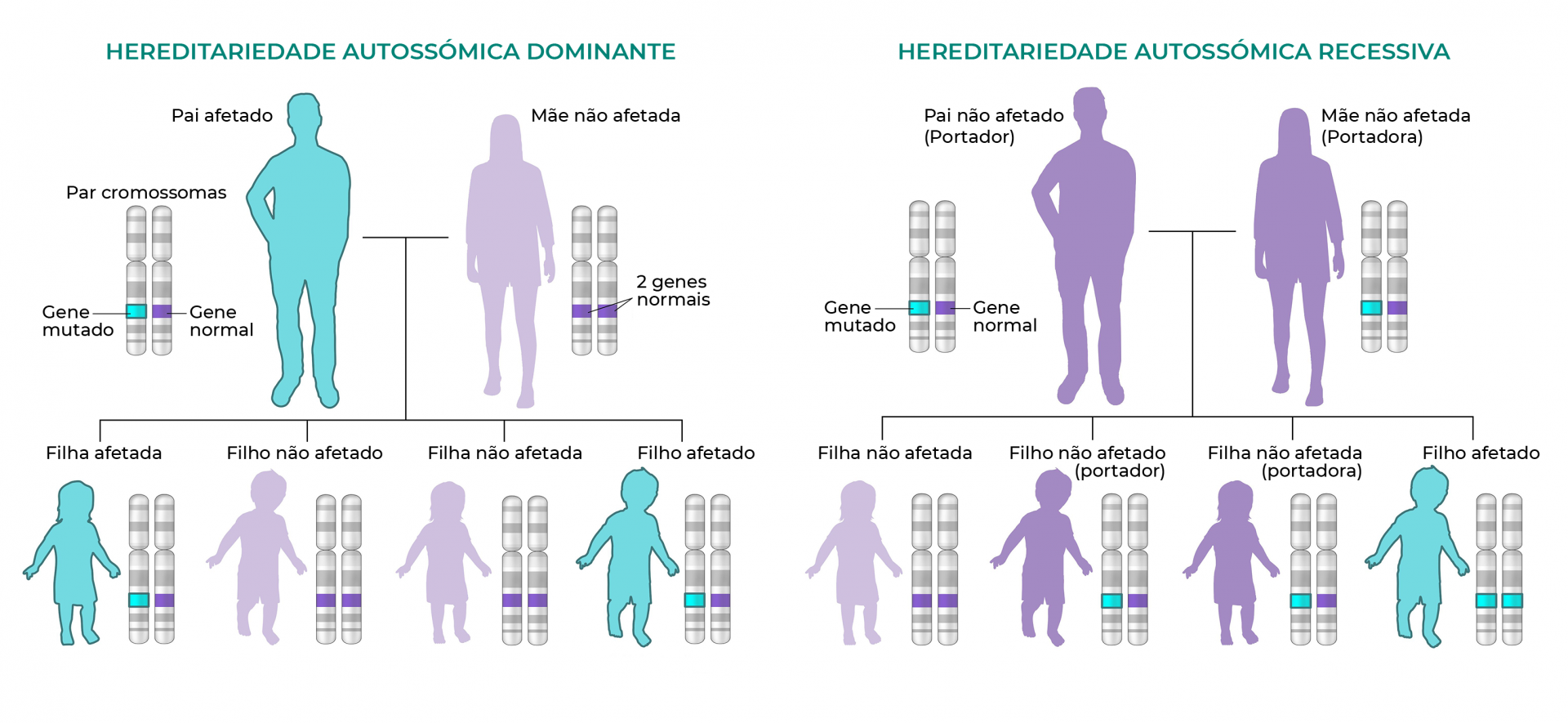
A displasia diastrófica é uma condição genética rara herdada num padrão autossómico recessivo, o que significa que ambas as cópias do gene em cada célula têm mutações. Os pais de um bebé com um padrão autossómico recessivo transportam, cada um, uma cópia do gene mutado, mas normalmente não mostram sinais e sintomas da condição3. As pessoas com esta condição apresentam baixa estatura e braços e pernas muito curtos. A maioria tem também dores articulares precoces (osteoartrite) e deformidades nas articulações, chamadas contraturas, que restringem o movimento.
{kind=link}
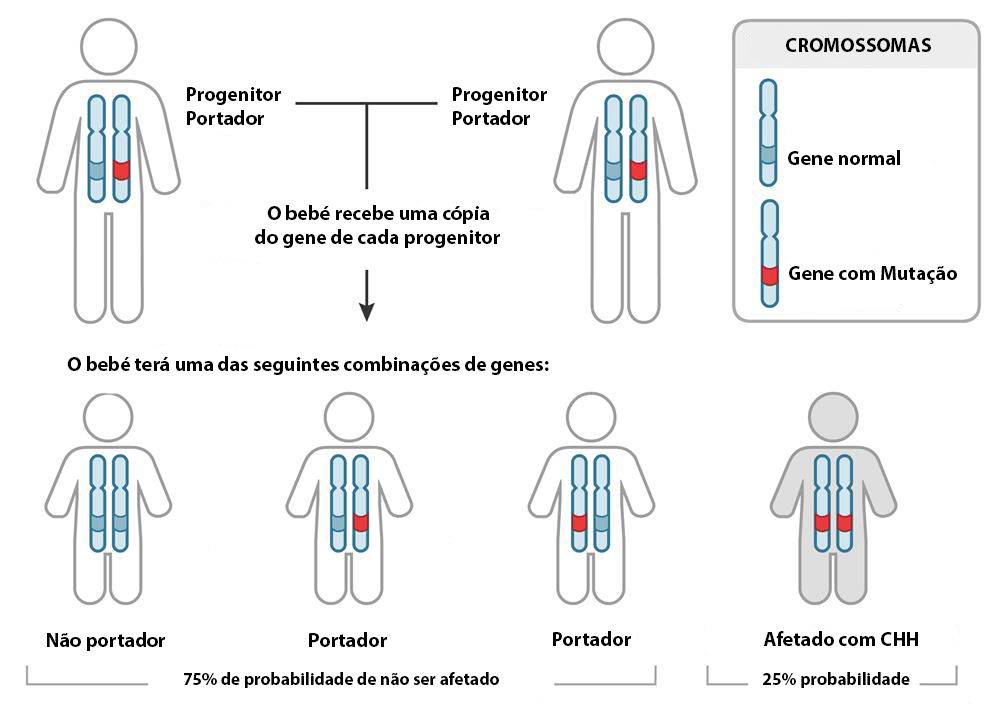
Fig. 2. Hereditariedade autossómica recessiva.
O que causa a Displasia Diastrófica4?
A displasia diastrófica é uma das várias displasias ósseas causadas por alterações de sulfatação resultantes de mutações no gene SLC26A2. Este gene fornece instruções para fazer a proteína SLC26A2 que transporta os iões de sulfato essenciais para a formação normal da cartilagem e posterior conversão em osso.
A maioria da cartilagem é convertida em osso, excepto a cartilagem que continua a cobrir e proteger as extremidades dos ossos e que está presente no nariz e nas orelhas. As mutações no gene SLC26A2 alteram a estrutura da cartilagem em desenvolvimento, impedindo a formação adequada do osso e resultando nos problemas caraterísticos da displasia diastrófica4.
Características Clínicas e Radiológicas4,5
Características Clínicas5
|
Encurtamento dos membros |
Deslocamento do raio |
|
Crânio de tamanho normal |
Fenda palatina (em cerca de 1/3 dos bebés) |
|
Encurtamento ligeiro do tronco |
Inchaço do ouvido no período neonatal (em cerca 2/3 de bebés) |
|
Polegares de boleia (Figura 1) |
Desvio ulnar dos dedos das mãos |
|
Peito pequeno |
Espaço entre o primeiro e o segundo dedo do pé |
|
Abdómen proeminente |
Pé torto |
|
Contracturas das grandes articulações |
Hemangiomas planos na testa |

|
Fig.3. Mão de bebé com displasia diastrófica. Braquidactilia |
Características Radiográficas
|
Crânio de tamanho normal com um esqueleto curto desproporcionado |
Hipoplasia das ilíacas com acetábulos planos; |
|
Cifose cervical presente na maioria dos bebés e crianças |
Os ossos longos surgem moderadamente encurtados |
|
Ossificação das vértebras torácicas superiores incompleta |
Úmero distal por vezes bífido ou em V, pontiagudo e hipoplástico |
|
Fissuras coronais presentes nas vértebras lombares e torácicas inferiores |
O fémur é distalmente arredondado |
|
Estreitamento da distância interpedicular de L1 a L5 frequente |
Patela fragmentada ou com várias camadas |
|
Costelas curtas e peito em forma de sino |
Raio e tíbia podem ser curvados |
|
Esterno pode apresentar duplicação dos centros de ossificação |
Luxação radial proximal pode estar presente no nascimento |
As mãos podem também apresentar características típicas (Figuras 3 e 4):
- Braquidactilia
- Abdução bilateral do polegar com desvio ulnar dos dedos
- Ausência do primeiro metacarpo
- Falanges proximais e médias em forma de delta
- Em alguns casos graves, ossificação de dois a três ossos do carpo no recém-nascido, simulando uma idade esquelética avançada

|
Fig 4. Bebé. A - Extremidades inferiores: rizomelia, mesomelia. |
Diagnóstico2,3
|
Quando está presente um historial familiar claro: |
|
Em algumas famílias com antecedentes de displasia diastrófica é possível um diagnóstico antes do nascimento (pré-natal), durante o início da gravidez com base nos resultados de testes genéticos especializados |
|
Por vezes pode ser detectada durante o segundo trimestre através de ultra-sonografia fetal, uma técnica de imagem especializada na qual são utilizadas ondas sonoras para criar uma imagem do feto em desenvolvimento |
|
Achados característicos durante a ultra-sonografia fetal: |
|
Encurtamento marcado dos ossos dos dedos (falanges), braços e pernas |
|
Desvio anormal dos polegares e dedos dos pés grandes |
|
Deformidades graves de ambos os pés (talipes ou "pés tortos"). Fig. 2D |
Na maioria dos casos, a displasia diastrófica é diagnosticada e/ou confirmada à nascença com base numa avaliação clínica completa, na identificação dos achados físicos caraterísticos referidos e numa variedade de testes especializados, tais como técnicas avançadas de imagem: TAC, RM, etc. Podem também ser realizados testes audiológicos para ajudar a detectar défices auditivos que podem ocorrer em algumas crianças.
Diagnóstico DIferencial2,3
Os sinais e sintomas da displasia diastrófica são semelhantes aos de outra displasia óssea chamada atelosteogénese tipo 2 (também conhecida como displasia óssea neonatal I, deriva de mutações no mesmo gene que causa a displasia diastrófica). Contudo, esta tende a ser menos grave e, embora alguns bebés afectados tenham problemas respiratórios, a maioria das pessoas com displasia diastrófica vive até à idade adulta. Outras condições devem ser descartadas para chegar ao disgnóstico:
- Acondrogénese tipo IB é outra doença genética rara que também se pensa ser causada por mutações do gene SLC26A2.
- Displasia pseudodiastrófica é uma doença genética rara também caracterizada por braços e pernas curtos, baixa estatura e deformidades graves dos pés.
- Artrogripose múltipla congénita é um grupo de condições congénitas que se caraterizam por movimento limitado ou imobilidade de várias articulações e substituição parcial ou completa do músculo por tecido fibroso nas áreas afetadas.
- Displasia epifisária múltipla recessiva é caracterizada por dores nas articulações, deformidades das mãos, pés e joelhos e escoliose.
Tratamento2
O tratamento da displasia diastrófica é direcionado para os sintomas específicos de cada pessoa e pode exigir os cuidados coordenados de uma equipa de especialistas: pediatras, ortopedistas, cirurgiões, fisioterapeutas, ortodontistas, audiologistas e outros profissionais de saúde.
Os médicos devem monitorizar os bebés para assegurar detecção imediata e tratamento preventivo ou corretivo adequado. Podem ainda ser utilizadas medidas especiais de apoio para ajudar a assegurar uma ingestão adequada de nutrientes em bebés com fissura palatina.
Em alguns casos podem ser realizados procedimentos cirúrgicos para corrigir malformações que resultem em dificuldades respiratórias e/ou de alimentação. A fisioterapia em combinação com medidas cirúrgicas e de apoio pode ser útil para melhorar a capacidade de caminhar e realizar outros movimentos.
De acordo com a literatura médica existente, embora as deformidades do pé associadas a esta condição possam ser resistentes ao tratamento, a terapia precoce e persistente pode ser útil na obtenção de resultados benéficos.
Em crianças com problemas dentários, poderão ser necessários aparelhos ortodônticos, cirurgia e/ou outros procedimentos corretivos. Por fim, as injecções de esteróides podem também ser utilizadas para ajudar a diminuir deformidades do ouvido que frequentemente afetam os bebés.
Aconselhamento Genético
O aconselhamento genético será benéfico para as pessoas afetadas e as suas famílias, pois fornece informações sobre a natureza, hereditariedade e implicações das doenças genéticas, ajudando a tomar decisões médicas e pessoais informadas, essenciais, por exemplo, ao nível do planeamento familiar, onde se deve esclarecer sobre o risco genético e a possibilidade de testes pré-natais.
É apropriado oferecer aconselhamento genético (incluindo a discussão de potenciais riscos para os descendentes e opções reprodutivas) aos jovens adultos que são afetados.
Bibliografia
1. C.M. McNamara, et al. Cleidocranial dysplasia: radiological appearances on dental panoramic radiography Dentomaxillofac Radiol, 28 (1999), pp. 89-97.
2. Bruderer M, et al. Role and regulation of RUNX2 in osteogenesis. Eur Cell Mater. 2014 Oct 23;28:269-86. doi: 10.22203/ecm.v028a19. PMID: 25340806.
3. Chin-Yun Pan, et al. Craniofacial features of cleidocranial dysplasia,Journal of Dental Sciences,Volume 12, Issue 4,2017,Pages 313-318,ISSN 1991-7902,https://doi.org/10.1016/j.jds.2017.07.002.
4. Farrow E, et al. Cleidocranial Dysplasia: A Review of Clinical, Radiological, Genetic Implications and a Guidelines Proposal. J Craniofac Surg. 2018 Mar;29(2):382-389. doi: 10.1097/SCS.0000000000004200. PMID: 29189406.
5. López, B. et al. Cleidocranial Dysplasia: report of a family, Journal of Oral Science, Volume 46, Número 4, (2004) pp. 259-266
6. Kreiborg, S, Jensen, BL. Tooth formation and eruption – lessons learnt from cleidocranial dysplasia. Eur J Oral Sci 2018; 126(Suppl. 1): 72– 80. © 2018 Eur J Oral Sci
7. Jensen, B.L. and Kreiborg, S. (1990), Development of the dentition in cleidocranial dysplasia. Journal of Oral Pathology & Medicine, 19: 89-93. https://doi.org/10.1111/j.1600-0714.1990.tb00803.
Saiba mais sobre:
-
O processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
-
A Dor nas displasias ósseas aqui