
Displasia Espondiloepifisária Congénita - SEDC
A displasia espondiloepifisária congénita (SEDc) é uma displasia óssea rara com incidência ou prevalência exacta desconhecida. No entanto, Estima-se que ocorra em aproximadamente 1 em cada 100.000 nascimentos1.
O que é a SEDc1?
A SEDc é uma condrodisplasia rara autossómica dominante que pertence ao grupo das displasias ósseas com origem em mutações no gene COL2A1. Apresenta estatura baixa e desproporcionada (tronco curto), epífises anormais, e corpos vertebrais achatados. As características esqueléticas manifestam-se ao nascimento e evoluem com o tempo. Outras características incluem a miopia e/ou degeneração da retina com descolamento da retina e fenda palatina1.
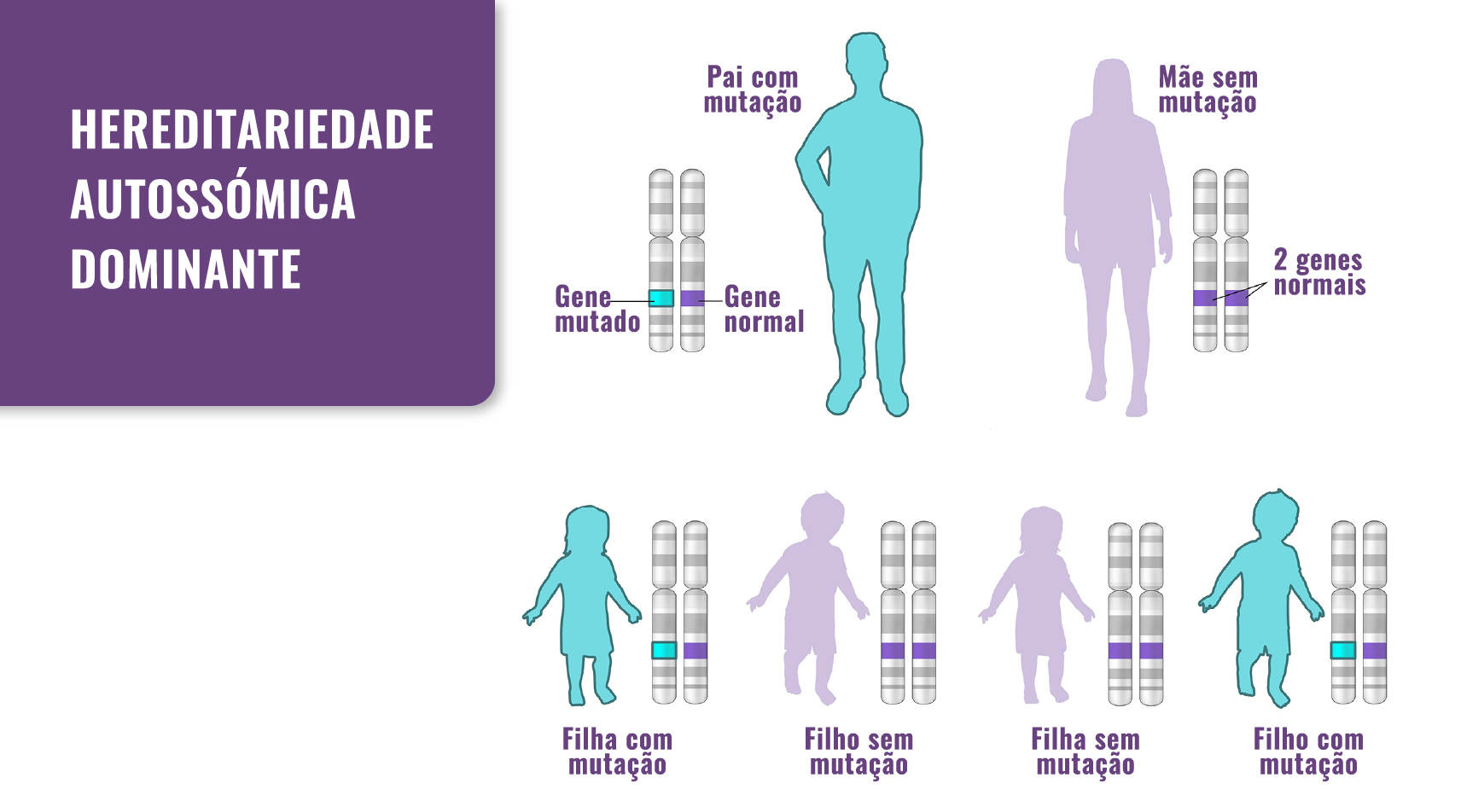
Fig. 1. Hereditariedade: a SEDc passa de pais para filhos. No entanto, também pode ocorrer como resultado de uma mutação de novo.
O que causa a SEDc1?
A SEDc é causada por uma mutação no gene COL2A1. Os genes fornecem instruções para a criação de proteínas que desempenham um papel crítico em muitas funções do corpo. Quando ocorre uma mutação de um gene, o seu produto proteico pode ser defeituoso, ineficiente, ou ausente e, dependendo das funções da proteína em particular, isto pode afectar muitos sistemas de órgãos do corpo. O gene anormal pode ser herdado de qualquer dos pais (autossómico), ou pode ser o resultado de uma nova mutação no indivíduo afectado (mutação de novo). O risco de passar o gene anormal do progenitor afectado para os descendentes é de 50%, independentemente do sexo da criança.
O gene COL2A1 contém instruções para a criação (codificação) de colagénio tipo II, uma das proteínas mais abundantes no corpo e um importante bloco de construção de tecido conector (o material entre as células do corpo que dá forma e força aos tecidos). Existem muitos tipos diferentes de colagénio e o colagénio tipo II é mais predominante na cartilagem e no fluido gelatinoso que preenche o centro dos olhos (humor vítreo), mas também se encontra no osso. As mutações do gene COL2A1 resultam em níveis diminuídos de colagénio tipo II funcional, que acabam por levar a um crescimento anormal do esqueleto.
Características Clínicas e Radiológicas2,3
As características manifestam-se ao nascimento, com a ossificação tardia das epífises como a sua marca distintiva. A altura dos adultos varia geralmente entre 84-128 cm. Esta condição apresenta um conjunto de características clínicas e radiográficas essenciais:
-
Vértebras bulbosas e em forma de pêra à nascença que mais tarde achatam
-
Platispondilia severa com finos espaços de discos intervertebrais (Figura 1A, B)
-
Cifoscoliose, lordose lombar e instabilidade atlanto-axial. A instabilidade atlanto-axial é secundária à hipoplasia odontóide e, subsequentemente, confere um risco muito maior de mielopatia cervical
-
Ausência de ossos púbicos ao nascimento com telhados horizontais de acetábulos e asas ilíacas curtas e largas
-
Ausência de epífises de calcâneo e joelho à nascença
-
Mais tarde, há atraso na ossificação das cabeças do fémur (Figura 1C)
-
Observa-se ossificação tardia do carpo e do tarso, mas o desenvolvimento das mãos e pés normalmente não é afetado
-
Crânio grande e dolicocefálico (Figura 1D)
-
Encurtamento rizomélico das extremidades, mais nos membros inferiores do que nos superiores e alargamento metafisário secundário às epífises anormais (Figura 1E e F).
 |
|
Fig. 2. Radiografias laterais da espinha dorsolombar mostram platispondilia (seta, A) com espaços de discos intervertebrais gravemente reduzidos (seta, B). A radiografia da pélvis (C) mostra pequenas epífises femorais (seta branca), acetábulos horizontais (seta preta) e asas ilíacas curtas (a). A radiografia do crânio (D) mostra calvário relativamente aumentado (seta). As radiografias de membros inferiores (E, F) mostram fémures relativamente curtos e pequenas epífises com irregularidade metafisária secundária (seta, F). Fonte: World Jornal of Radiology |
Diagnóstico
O diagnóstico da SEDC baseia-se na identificação dos seus sintomas característicos, numa história detalhada do paciente, numa avaliação clínica exaustiva e numa variedade de testes especializados. À nascença poderá ocorrer suspeita, devido a descobertas características.
Testes e exames clínicos como radiografias podem ser utilizados para fornecer um exame minucioso e cuidadoso de todo o sistema ósseo (levantamento completo do esqueleto) a fim de detectar alterações no esqueleto que são características da SEDC.
Algumas técnicas de imagem mais avançadas, como a ressonância magnética (RM) e a tomografia computorizada (TAC) podem ser usadas para avaliar a saúde esquelética, particularmente antes da cirurgia para corrigir malformações esqueléticas. A RM utiliza um campo magnético e ondas de rádio para produzir imagens transversais de determinados órgãos e tecidos corporais, enquanto que a TAC cria um filme que mostra imagens transversais de certas estruturas de tecidos, recorrendo a um computador e aos raios X. É possível também recorrer a um exame de artrografia, no qual é injectado um corante nas ancas para avaliar a cartilagem.
Testes Genéticos
Os testes genéticos moleculares podem confirmar o diagnóstico ao detetar mutações no gene que causa a SEDC (COL2A1), mas está disponível apenas como um serviço de diagnóstico em laboratórios especializados.
Diagnóstico Diferencial4
Existem várias perturbações que ocorrem devido a uma mutação diferente no mesmo gene que causa a SEDC, sendo conhecidas como alélicas. Estas perturbações partilham frequentemente vários sintomas, que se sobrepõem uns aos outros, por isso as comparações podem ser úteis para um diagnóstico diferencial:
- Displasia de Kniest
- Síndrome de Stickler tipo I
- Acondrogénese tipo II
- Displasia otospondilomegaepifisária
- Displasia espondiloepimetafisiária tipo Strudwick
- Displasia espondiloepifisária tardia
- Síndrome de Mórquio
- Displasia metatrópica
Tratamento3
Não existe tratamento para a condição subjacente. O tratamento é direccionado para os sintomas específicos que se manifestam em cada indivíduo e pode exigir os cuidados coordenados de uma equipa de especialistas: pediatras, especialistas em diagnóstico e tratamento de perturbações músculo-esqueléticas (cirurgiões ortopédicos), especialistas em diagnóstico e tratamento de perturbações oculares (oftalmologistas), reumatologistas, fisioterapeutas e outros profissionais de saúde devem fazer um acompanhamento cuidadoso da criança afectada. O aconselhamento genético pode ser benéfico para os indivíduos afectados e as suas famílias. O apoio psicossocial para toda a família também é essencial.
As terapias específicas são sintomáticas e de apoio. Os médicos devem monitorizar cuidadosamente os bebés afectados para assegurar uma detecção imediata e uma prevenção ou tratamento correctivo adequado das dificuldades respiratórias (respiratórias).
Exames oftalmológicos regulares são necessários para detectar e avaliar a miopia e para prevenir o descolamento da retina. O descolamento da retina pode ser reparado cirurgicamente. A fisioterapia pode melhorar o movimento articular e evitar a degeneração muscular (atrofia). Os procedimentos cirúrgicos podem ser recomendados para fechar uma fenda palatina.
Certas características físicas associadas à SEDC, especificamente pescoço curto, instabilidade da coluna cervical, capacidade pulmonar reduzida e pequenas vias respiratórias podem complicar o uso da anestesia. Os indivíduos afectados precisam de ser avaliados antes de serem submetidos a procedimentos que exijam anestesia.
O tratamento da SEDC varia consoante as condições ortopédicas associadas e pode incluir:
-
Fusão cervical e possível descompressão da coluna cervical
-
Pode ser necessário auréola e cinta ou colete de suporte para a escoliose e cifose, se detetada cedo
-
Fusão vertebral para escoliose e cifose ou implantação de uma haste que trata a deformidade da coluna vertebral na criança, mas permite o crescimento contínuo e controlado da coluna vertebral
-
Osteotomia das ancas para corrigir desalinhamento e/ou subluxação (luxação parcial e contraturas de flexão da anca). Em alguns casos pode ser necessária cirurgia de substituição total da anca (artroplastia total da anca)
-
Fundição para deformidades dos pés.
Bibliografia
Referências
1. Anderson, I., et al. Spondyloepiphyseal dysplasia congenita: genetic linkage to type II collagen (COL2A1). Am. J. Hum. Genet. 46: 896-901, 1990.
2. McKay SD, et al. Review of cervical spine anomalies in genetic syndromes. Spine (Phila Pa 1976). 2012;37:E269-E277.
3. Veeravagu A, et al. Neurosurgical interventions for spondyloepiphyseal dysplasia congenita: clinical presentation and assessment of the literature. World Neurosurg. 2013;80:437.e1-8.
4. Fraser, G. et al. Dysplasia spondyloepiphysaria congenita and related generalized skeletal dysplasias among children with severe visual handicaps. Arch. Dis. Child. 44: 490-498, 1969.
Outras Fontes:
Saiba mais sobre:
-
O processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
-
A Dor nas displasias ósseas aqui
-
doenças raras e displasias ósseas através dos nossos vídeos informativos