
Displasia Cleidocraniana
A displasia cleidocraniana (DCC) é uma condição genética rara, também conhecida como síndrome de Scheuthauer-Marie-Sainton ou disostose cleidocraniana, que se caracteriza por um desenvolvimento anómalo do esqueleto e dos dentes. A sua prevalência é estimada em 1 nascimento por milhão1.
O que é a Displasia Cleidocraniana1?
A DCC é uma condição genética rara que afeta os dentes e os ossos, como o crânio, a face, a coluna vertebral, as clavículas e as pernas. Os ossos das pessoas com DCC podem ter uma formação diferente ou ser mais frágeis do que o normal, e alguns ossos, como as clavículas, podem estar ausentes. As pessoas diagnosticadas com DCC podem ter características físicas distintas, como clavículas subdesenvolvidas, baixa estatura, características faciais únicas e atraso no desenvolvimento dos dentes.
O termo cleidocraniana deriva do grego cleido (clavícula) e kranion (cabeça).
O que causa a Displasia Cleidocraniana1,2?
Na maioria dos casos a transmissão é autossómica dominante, o que significa que um dos progenitores transmite a condição para a criança com uma probabilidade de 50%. Em 20-40% dos casos registados ocorre de forma espontânea.
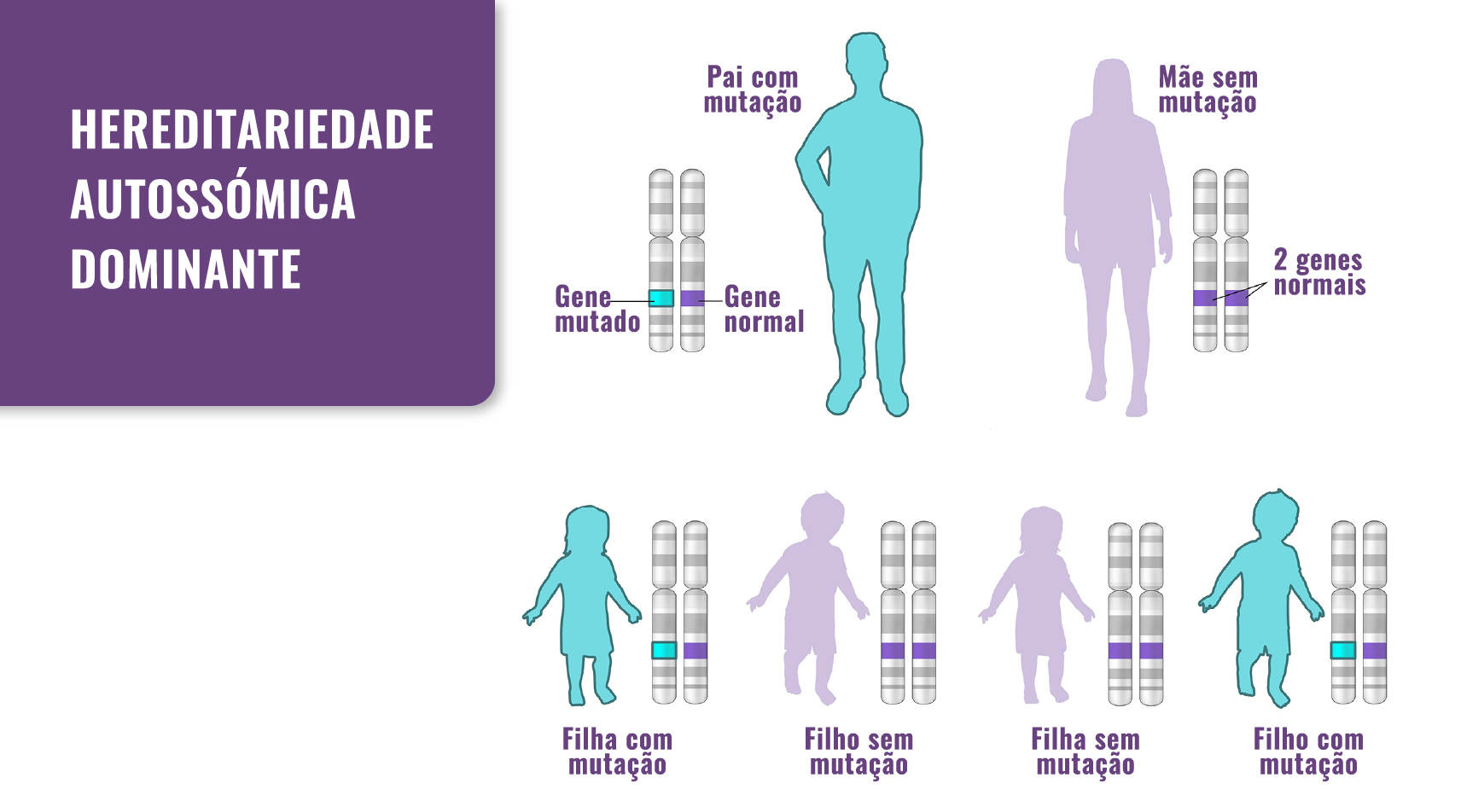
Fig. 1. Hereditariedade: a DCC passa de pais para filhos. No entanto, também pode ocorrer como resultado de uma mutação de novo.
É geralmente causada por mutações no gene RUNX2. Este gene fornece instruções para a produção de uma proteína que atua como um "interruptor" que regula uma série de outros genes envolvidos no desenvolvimento e manutenção dos ossos, dentes e cartilagens. As mutações neste gene reduzem ou eliminam a atividade da proteína produzida, interferindo com o desenvolvimento normal dos ossos, dentes e cartilagens.
Em cerca de 30% das pessoas com DCC, não foi encontrada qualquer mutação no gene RUNX2. A causa da condição nestes indivíduos é ainda desconhecida.
Características Clínicas e Radiológicas3,4
Características Clínicas — Esqueléticas
- Clavículas subdesenvolvidas ou ausentes. As pessoas com DCC apresentam geralmente clavículas subdesenvolvidas ou ausentes. Como resultado, os seus ombros são estreitos e inclinados e podem estar invulgarmente aproximados em frente do corpo.
- A maturação tardia do crânio crânio provoca um atraso no fecho das linhas de crescimento onde os ossos do crânio se encontram (suturas).
- Presença de espaços maiores que o normal (fontanelas) entre os ossos do crânio que são percetíveis como "pontos moles" na cabeça do bebé. As fontanelas fecham-se na primeira infância, mas podem permanecer abertas durante toda a vida em pessoas com esta condição;
-
Algumas pessoas apresentam pedaços extra de osso chamados ossos Wormian dentro das suturas;

|
Fig. 2 - Tomografia computorizada de rapaz de 7 anos com DCC. Mostra ossificação retardada dos ossos do crânio, sutura aberta, hipoplasia dos ossos nasais e múltiplos ossos Wormian. Fonte: Kreiborg, S. |
-
Baixa estatura. As pessoas afetadas são frequentemente mais baixas do que a média da população geral;
-
Dedos curtos e afilados e polegares largos;
-
Pés chatos;
-
Genu valgum. Joelhos que dobram para dentro;
-
Omoplatas curtas;
-
Escoliose. Curvatura anormal da coluna vertebral;
-
As mulheres têm um risco acrescido de necessitarem de uma cesariana no parto, devido a uma pélvis estreita que impede a passagem da cabeça do bebé.
Características faciais:
-
Braquicefalia. Crânio largo e curto;
-
Testa proeminente;
-
Hipertelorismo. Olhos largos e afastados;
-
Hipoplasia dos ossos nasais;
-
Maxilar superior pequeno.
Densidade dos ossos:
-
Osteopenia. Densidade óssea reduzida;
-
Podem desenvolver osteoporose, uma condição que torna os ossos progressivamente mais frágeis e propensos à fractura, numa idade relativamente precoce.
Características Clínicas — Anomalias dentárias3,4
-
São muito comuns e podem incluir perda retardada dos dentes decíduos (bebé);
-
Atraso na erupção dos dentes permanentes (adultos);
-
Dentes com forma invulgar, semelhantes a pinos;
-
Desalinhamento dos dentes e maxilares (má oclusão);
-
Formação de vários dentes permanentes supranumerários (extra), por vezes acompanhados de quistos nas gengivas;
-
Raízes ou coroas dentárias anómalas;
-
Palato muito arqueado ou fenda palatina.
Outras características
Para além das anomalias esqueléticas e dentárias, as pessoas com DCC podem ter perda de audição e são propensas a infecções sinusais e dos ouvidos4. Alguns bebés podem apresentar ligeiro atraso no desenvolvimento de capacidades motoras, tais como rastejar e andar, mas a inteligência não é afectada3.
Diagnóstico5
O diagnóstico pode ser feito numa fase intrauterina através de um ultrassom. Esta condição é diagnosticada quando as principais características da doença são encontradas durante um exame clínico e nas radiografias ao crânio e ao tórax. A realização de uma análise genética molecular também é necessária, sendo que esta é imprescindível para a realização do diagnóstico precoce5.
Tratamento6,7
Os tratamentos dependem de como a condição afeta cada pessoa. Existem múltiplos procedimentos ortopédicos, reconstrutivos faciais e dentários para melhorar o conforto, função e bem-estar. Os tratamentos recomendados podem incluir:
-
Nas crianças, cirurgia reconstrutiva facial nos ossos do rosto para remodelar a testa ou as maçãs do rosto
-
Procedimentos de fusão vertebral para apoiar a coluna vertebral
-
Cirurgia na parte inferior das pernas para corrigir os joelhos
-
Reparação cirúrgica de fracturas devidas a ossos frágeis
-
Remoção de pequenos pedaços de clavícula que podem afectar o plexo braquial e causar dores no braço ou problemas nervosos
-
Tubos para tratar infecções dos ouvidos
-
Suplementos de cálcio e vitamina D para fortalecer os ossos
Tratamento de Problemas Dentários7
Quase todas as pessoas com esta condição apresentam anomalia dentária e estes problemas podem afetar a oclusão, ou seja, o modo como os dentes superiores e inferiores se encaixam quando a boca está fechada.
A ortodontia e a cirurgia oral podem ajudar. O primeiro passo é uma avaliação cuidadosa de cada dente, seguida da extracção de qualquer dente extra. Para manter dentes viáveis, pode-se remover tecido gengival e orientar o crescimento dos dentes com recurso a aparelho ortodôntico. Para um resultado mais rápido, algumas pessoas preferem implantes, pontes ou próteses dentárias. Por vezes, é necessária uma cirurgia ao maxilar para corrigir a oclusão.
 |
|
Fig. 3 - Fotografias intra-orais de um rapaz com erupção espontânea dos incisivos definitivos após extracção dos incisivos primários, remoção cirúrgica dos incisivos supranumerários e cobertura óssea dos primeiros incisivos permanentes, formados aos 9,5 anos de idade. A fotografia A foi obtida aos 10 anos de idade e a fotografia B aos 11 anos. Fonte: Kreiborg, S. Tooth formation and eruption – lessons learnt from cleidocranial dysplasia. |
Os tratamentos exigem coordenação entre cirurgiões bucomaxilofaciais, ortodontistas e protesistas, bem como o dentista da família. Quanto mais cedo a doença for diagnosticada e tratada adequadamente, maiores serão as probabilidades de um resultado favorável.
|
Embora a DCC possa trazer vários desafios e exigir múltiplas cirurgias, as pessoas afetadas podem viver vidas longas e a alcançar os seus sonhos. |
 |
|
Gaten Matarazzo, o famoso ator da série Stranger Things tem DCC e realizou aos 17 anos a sua 4ª cirurgia para extração de dentes supranumerários (removeu 14 dentes) e para ajudar a expor 6 dentes que já deveriam ter crescido. Créditos: Netflix e Instagram do ator. |
Bibliografia
1. C.M. McNamara, et al. Cleidocranial dysplasia: radiological appearances on dental panoramic radiography Dentomaxillofac Radiol, 28 (1999), pp. 89-97.
2. Bruderer M, et al. Role and regulation of RUNX2 in osteogenesis. Eur Cell Mater. 2014 Oct 23;28:269-86. doi: 10.22203/ecm.v028a19. PMID: 25340806.
3. Chin-Yun Pan, et al. Craniofacial features of cleidocranial dysplasia,Journal of Dental Sciences,Volume 12, Issue 4,2017,Pages 313-318,ISSN 1991-7902,https://doi.org/10.1016/j.jds.2017.07.002.
4. Farrow E, et al. Cleidocranial Dysplasia: A Review of Clinical, Radiological, Genetic Implications and a Guidelines Proposal. J Craniofac Surg. 2018 Mar;29(2):382-389. doi: 10.1097/SCS.0000000000004200. PMID: 29189406.
5. López, B. et al. Cleidocranial Dysplasia: report of a family, Journal of Oral Science, Volume 46, Número 4, (2004) pp. 259-266
6. Kreiborg, S, Jensen, BL. Tooth formation and eruption – lessons learnt from cleidocranial dysplasia. Eur J Oral Sci 2018; 126(Suppl. 1): 72– 80. © 2018 Eur J Oral Sci
7. Jensen, B.L. and Kreiborg, S. (1990), Development of the dentition in cleidocranial dysplasia. Journal of Oral Pathology & Medicine, 19: 89-93. https://doi.org/10.1111/j.1600-0714.1990.tb00803.
A ANDO agradece a colaboração do Gabriel Lourenço (na foto) e da sua família na disponiblização da imagem para esta página.
Saiba mais sobre:
-
O processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
-
A Dor nas displasias ósseas aqui
-
Consulte uma apresentação sobre as limitações funcionais e sociais na pseudoacondroplasia aqui.